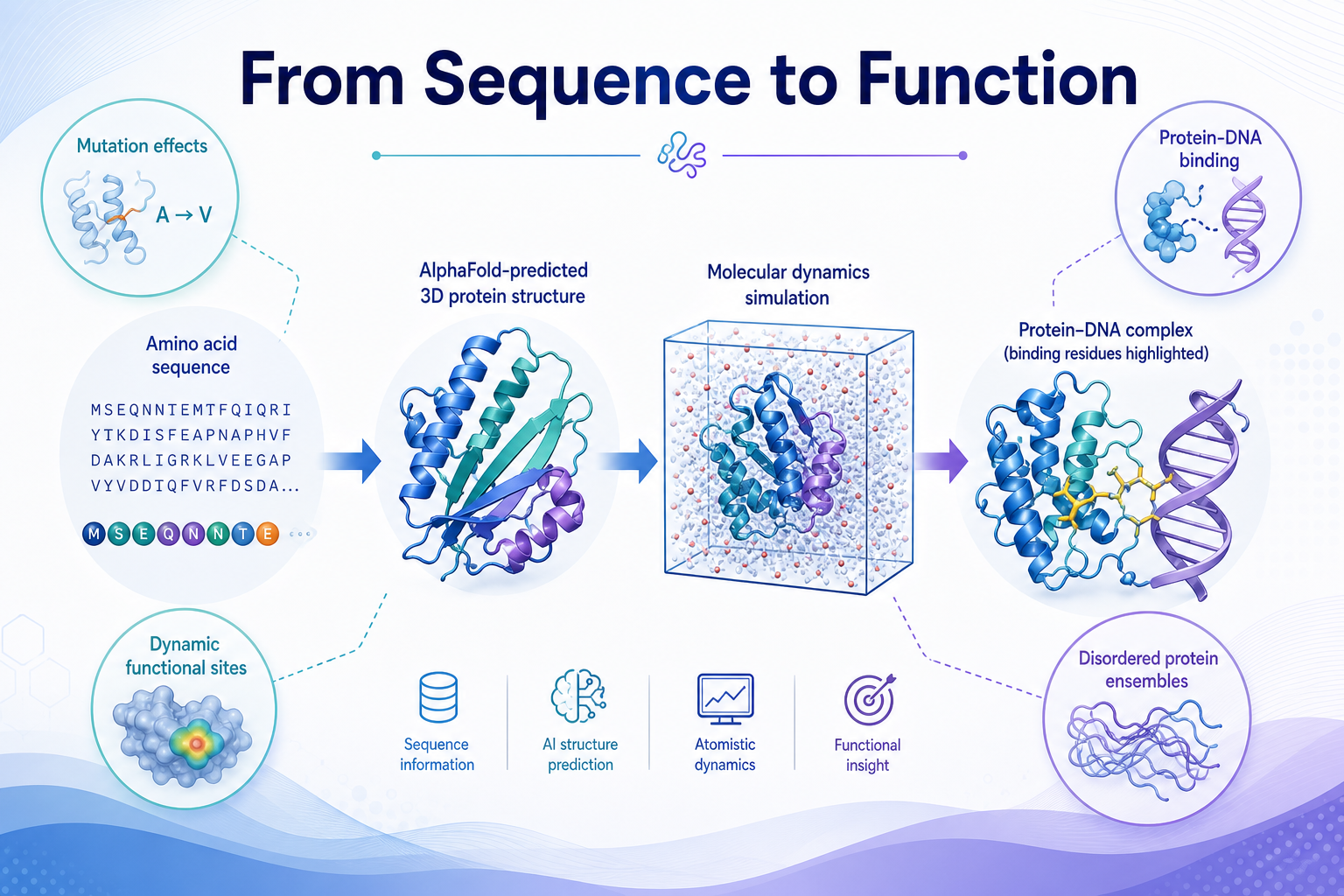
0導讀:為什麼要把 MD 和 AI 模型綁在一起
背景與動機
傳統的蛋白質結構預測模型(AlphaFold、RoseTTAFold、ESM 系列)主要針對序列與靜態結構訓練,能給出高精度的三維結構,但缺乏動態特性。對於內在無序區域、或缺乏同源序列的蛋白,它們在預測突變效應與功能上能力有限。
另一方面,分子動力學(Molecular Dynamics, MD)能從物理力場出發,描述構象變化、鍵結斷裂與側鏈運動,是捕捉蛋白動態的關鍵工具。把 MD 與深度學習模型結合,研究者既能享有 AI 模型的效率,又能補足靜態結構的盲點——尤其適用於預測突變對穩定性與結合親和力的影響。
1為什麼 AlphaFold 還不夠?
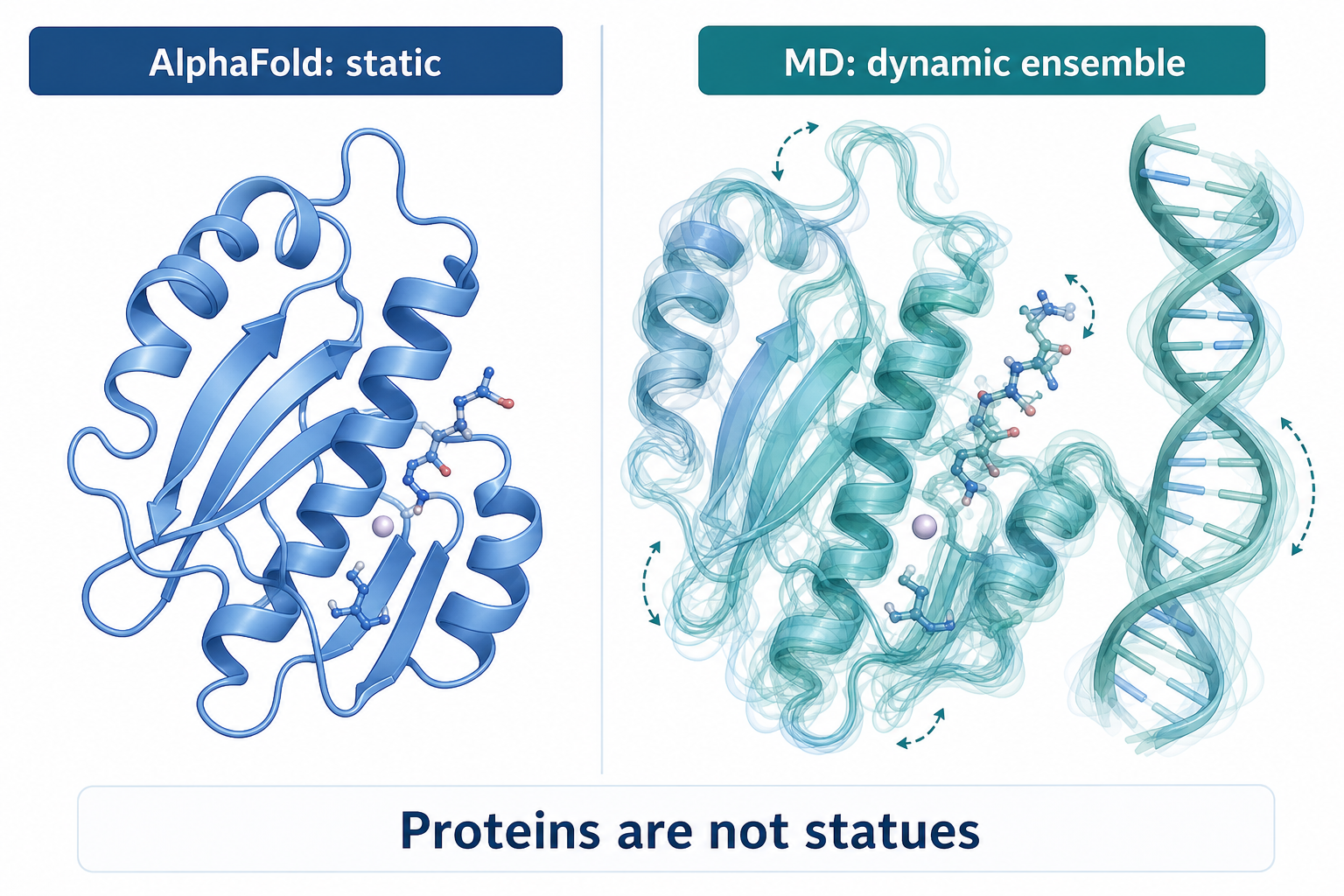
一句話:蛋白質不是雕像,它會動
AlphaFold 給你的是一張凍結的高解析快照——精準,但靜止。真實的蛋白質會擺動 loop、轉動側鏈、在結合 DNA 時局部變形。這些動態正是決定功能、酵素活性、以及突變後果的關鍵,而靜態結構看不到。MD 補上的就是這層「會動的構象集合」。
這張圖的生成提示詞(gpt-image-2, medium)
Split-screen scientific illustration comparing a static AlphaFold protein structure with a molecular dynamics simulation. Left: a single rigid protein ribbon, accurate but frozen like a high-resolution statue, label "AlphaFold: static". Right: the same protein under MD with multiple transparent overlapping conformations, moving loops, rotating side chains and a flexible DNA strand, label "MD: dynamic ensemble". Bottom caption "Proteins are not statues". Clean modern educational vector style, English labels only, soft blue teal white palette.
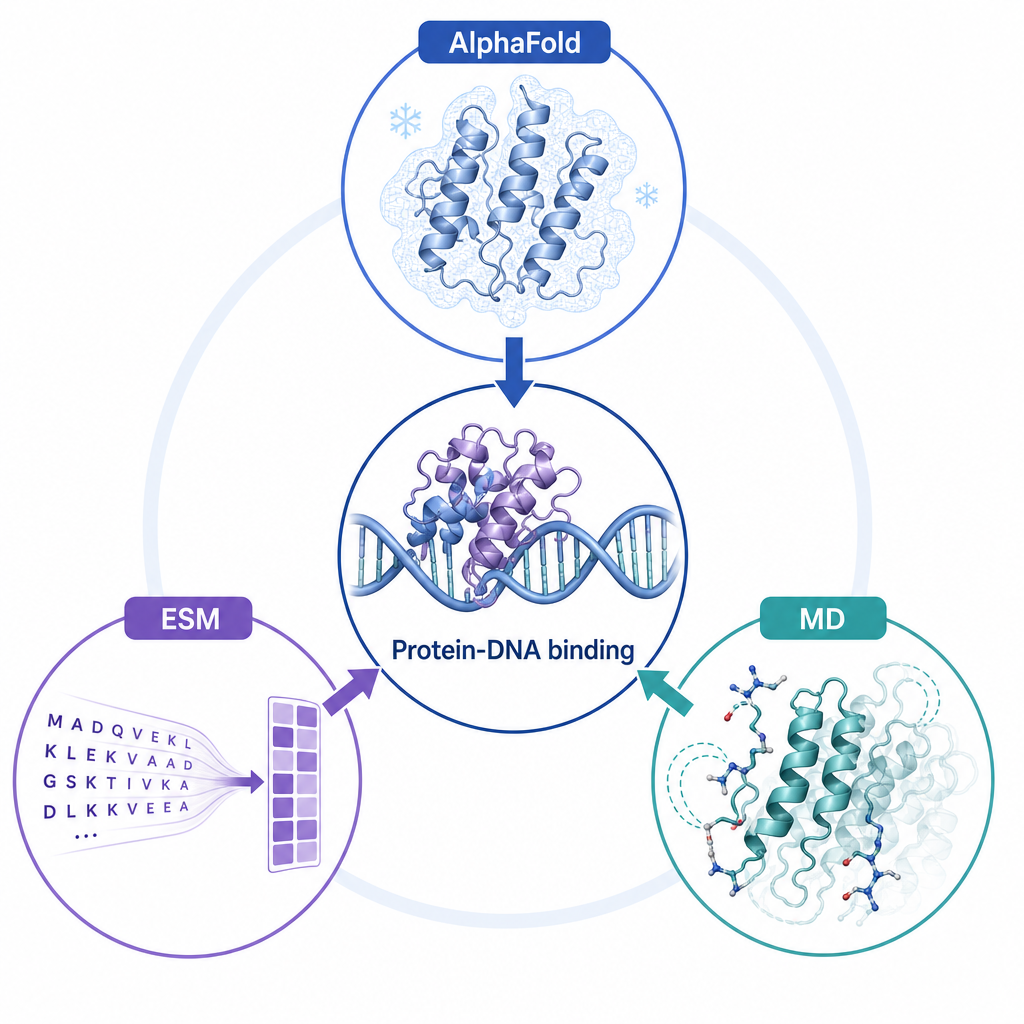
2三大方法如何互補
AlphaFold · ESM · MD 各補一塊拼圖
三種方法各自提供不同層次的資訊,最後匯聚到同一個問題上——蛋白–DNA 結合與突變效應:
- AlphaFold:序列 → 高精度靜態三維結構(提供起始模型)
- ESM:以蛋白語言模型從序列直接萃取演化與功能特徵
- MD:用物理力場補上動態運動(構象集合、柔性、接觸機率)
這張圖的生成提示詞(gpt-image-2, medium)
Educational infographic, triangular layout with three labeled nodes: top "AlphaFold" a frozen crystalline protein snapshot; bottom-left "ESM" amino acid letters transforming into protein features; bottom-right "MD" a protein with motion trails and flexible side chains. Center: a protein bound to a DNA double helix with three arrows converging. English labels only: AlphaFold, ESM, MD, Protein-DNA binding. Soft blue purple teal white palette, university teaching slide.
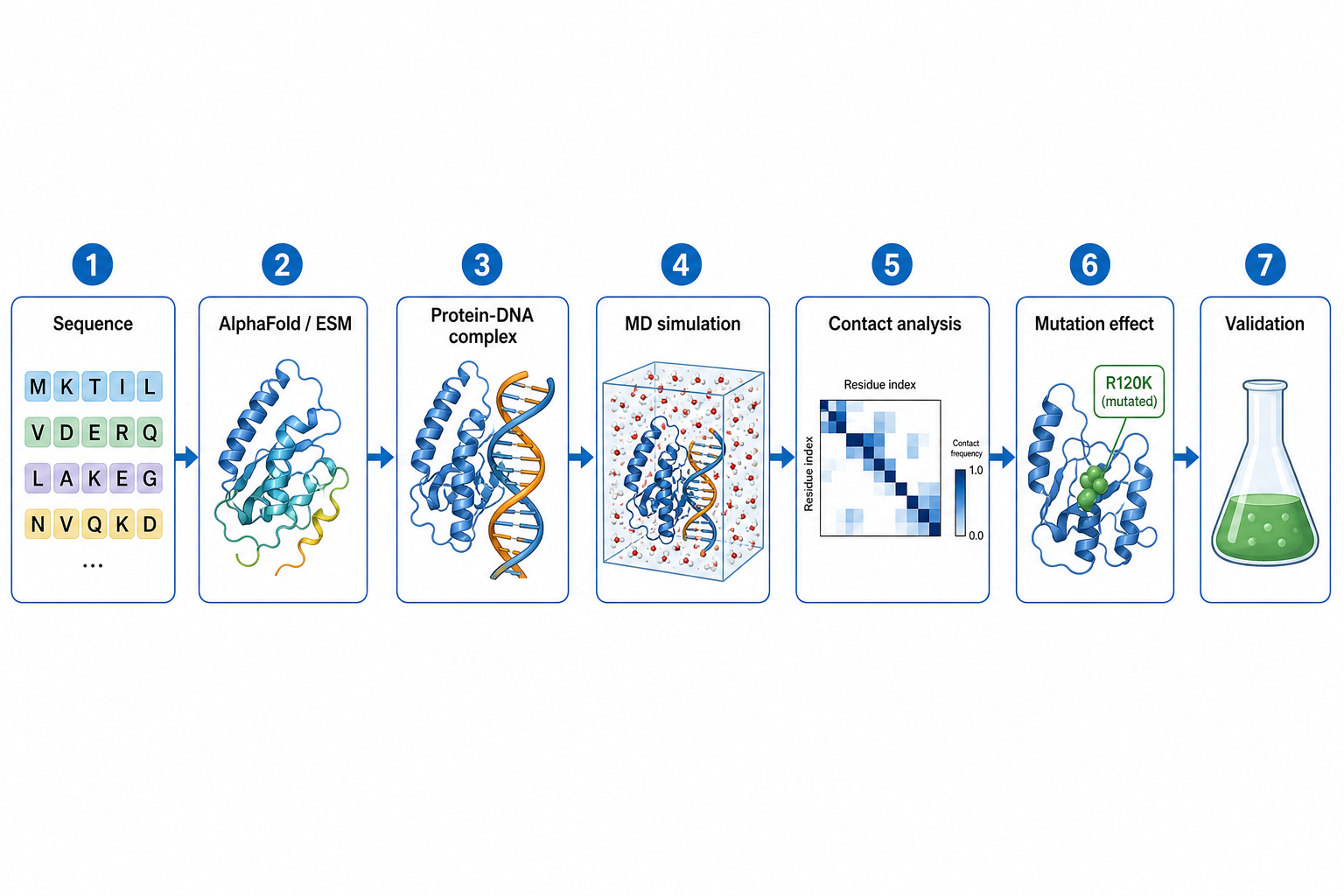
3完整研究流程
從一條序列,到突變效應與 DNA 結合的預測
一個典型的整合流程,由左到右串起 AI 結構預測與分子模擬:
這張圖的生成提示詞(gpt-image-2, medium)
Horizontal left-to-right workflow diagram, seven connected steps with icons and short English labels: 1 Sequence (letter blocks), 2 AlphaFold / ESM (folded protein), 3 Protein-DNA complex (protein + DNA helix), 4 MD simulation (simulation box with water), 5 Contact analysis (residue contact heatmap), 6 Mutation effect (highlighted mutated residue), 7 Validation (lab flask). Clean arrows, white background, blue and green accents, minimal English text, lecture slide.
4五個代表性研究
看真實論文怎麼把這套方法用出來
① SeqDance / ESMDance — 把 MD 動態餵進蛋白語言模型
研究者把 6 萬多個蛋白的 MD 模擬與法向模態分析所得的動態特徵(殘基移動相關性、溶劑可接觸表面積 SASA、二面角變化等)作為訓練標籤,開發出 SeqDance——它能直接從序列預測殘基層級的動態屬性,且其動態預測與實驗量得的折疊自由能變化(ΔΔG)顯著相關。
為利用「演化資訊」與「動態資訊」的互補,作者再把 SeqDance 的動態輸出與 ESM 結合,成為 ESMDance。
- 在 412 個蛋白上做 zero-shot 突變預測,ESMDance 的 Spearman 相關係數達 0.46
- 單獨用 ESM2 或 SeqDance 分別只有 0.33 與 0.24
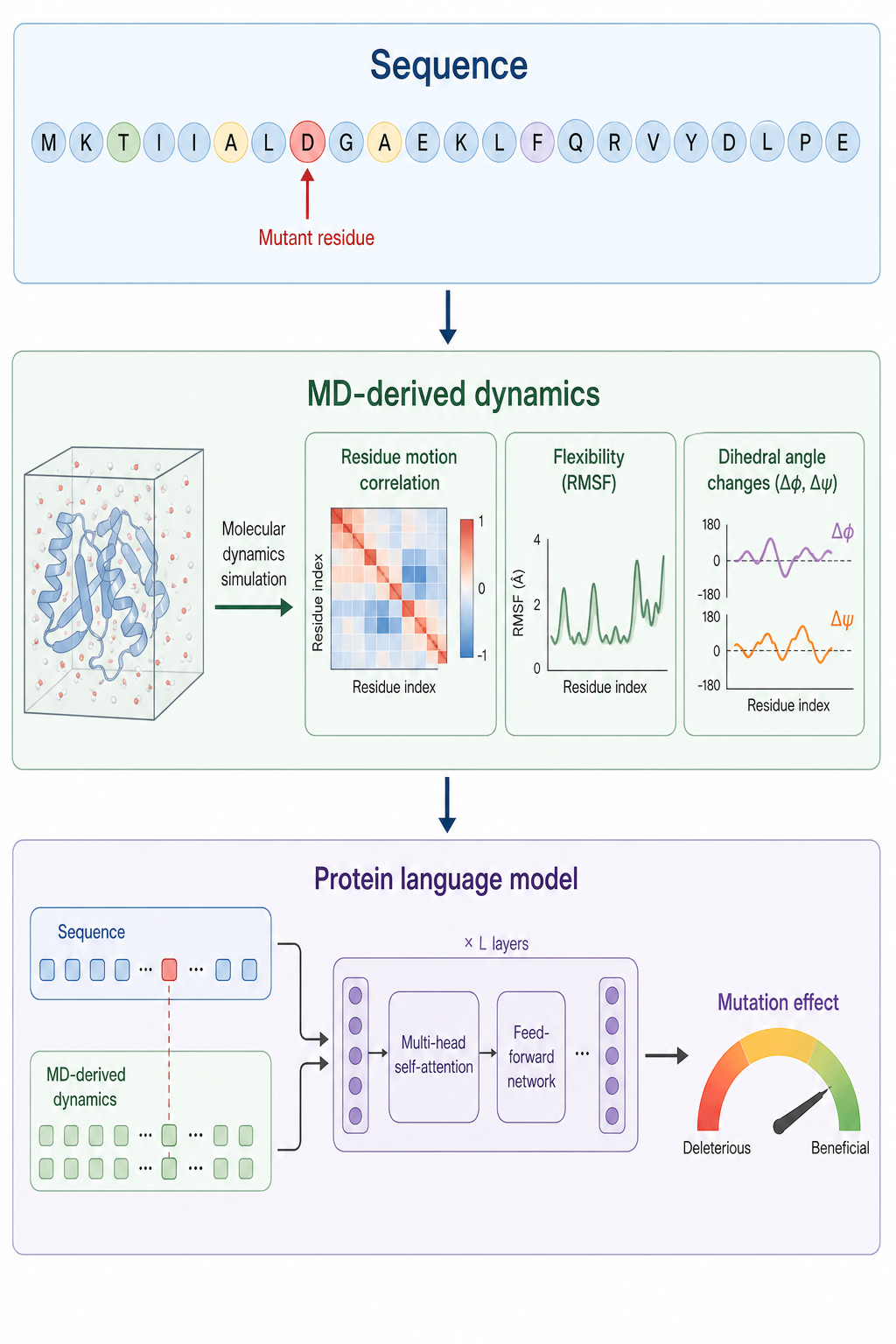
這張圖的生成提示詞(gpt-image-2, medium)
Three stacked horizontal layers. Top "Sequence" amino acid letters. Middle "MD-derived dynamics" an MD simulation producing residue motion correlation, flexibility, dihedral angle changes. Bottom "Protein language model" a neural network integrating sequence and dynamics, one mutant residue highlighted red, arrow to "Mutation effect". Flat vector style, soft colors, English labels only, bioinformatics education style.
② MELD-DNA — 用 MD + 貝葉斯推論預測 protein–DNA 複合體
在蛋白–DNA 結合領域,複合體的結構資料遠少於蛋白–蛋白複合體。MELD-DNA 利用貝葉斯推論,把 MD 模擬與有限的實驗或文獻資訊結合,以預測 protein–DNA 複合體結構。
它不僅能採樣多種結合模式,還能從構象聚類中挑出最有利的構象,並評估不同 DNA 序列之間的親和差異。
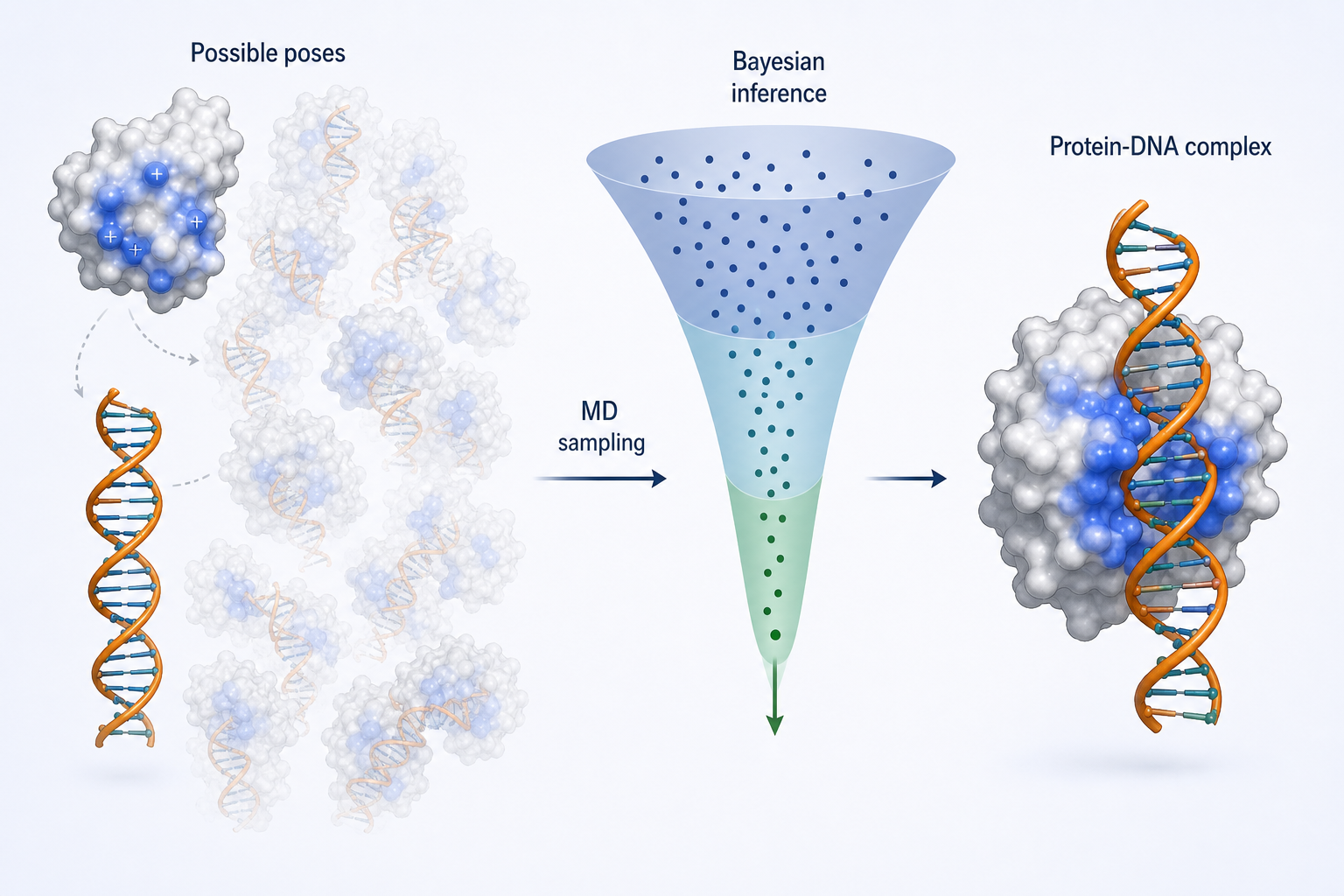
這張圖的生成提示詞(gpt-image-2, medium)
Conceptual illustration: left a protein with positive surface patches and a separate DNA helix surrounded by many faint transparent alternative binding poses; middle a downward narrowing funnel labeled "Bayesian inference"; right one final stable protein-DNA complex. English labels only: Possible poses, MD sampling, Bayesian inference, Protein-DNA complex. Elegant computational biology infographic, soft colors.
③ Cohesin — AlphaFold 補結構,再用 MD 找 DNA binding patch
人類 cohesin 複合體擁有長距離的 coiled-coil 區域,這些區域在實驗(cryo-EM)結構中常缺失。研究首先用 AlphaFold2 預測缺失的 coiled-coil 片段,再透過 MODELLER 把 cryo-EM 資料與 AF2 預測結合,建出全長 SMC1 / SMC3 子單元模型。
隨後在這些模型上跑粗粒化(coarse-grained)MD,用 DNA 與蛋白殘基的接觸概率與停留時間,辨識出多個 DNA binding patch。其中部分與已知突變研究一致,其他則是先前未報導的新位點。
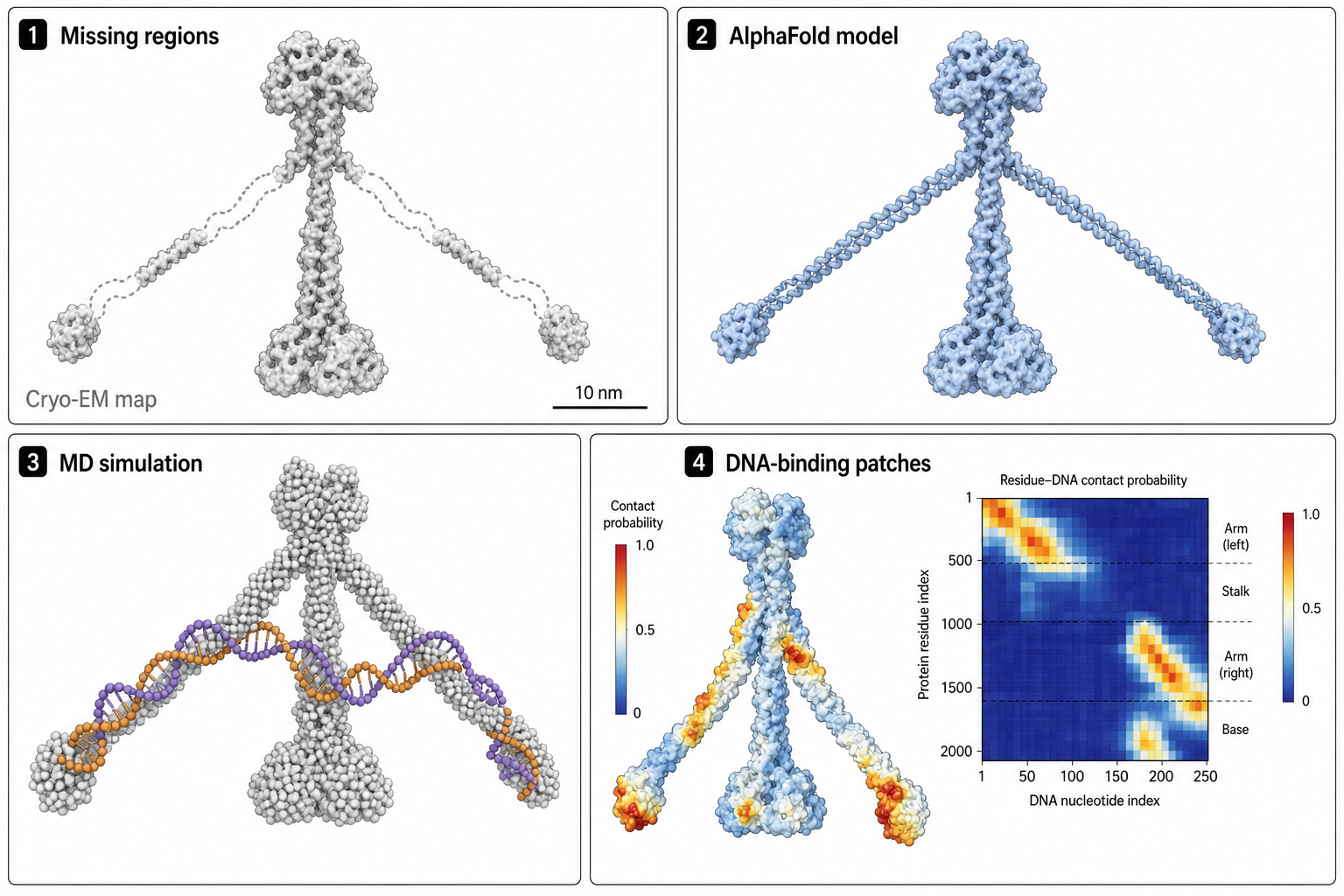
這張圖的生成提示詞(gpt-image-2, medium)
Four-panel case study of a large elongated protein complex. Panel 1 "Missing regions" cryo-EM structure with dashed gaps in long coiled-coil arms. Panel 2 "AlphaFold model" gaps filled in. Panel 3 "MD simulation" coarse-grained protein with a DNA strand. Panel 4 "DNA-binding patches" contact-probability heatmap. 2x2 grid, short English labels only, subtle colors, journal-club quality.
④ bAIes — 用 AlphaFold distogram 當先驗,描述無序蛋白
對於內在無序蛋白(IDPs),單一靜態結構難以描述其構象集合。bAIes 框架採用 AlphaFold2 輸出的殘基距離分佈(distogram)作為先驗(prior),結合物理力場進行貝葉斯抽樣,生成與 NMR 和 SAXS 實驗資料一致的原子分辨率構象集合。
作者指出,bAIes 在多個 IDPs 上產生的結構集合與全原子 MD 模擬相當,但計算成本大幅降低。
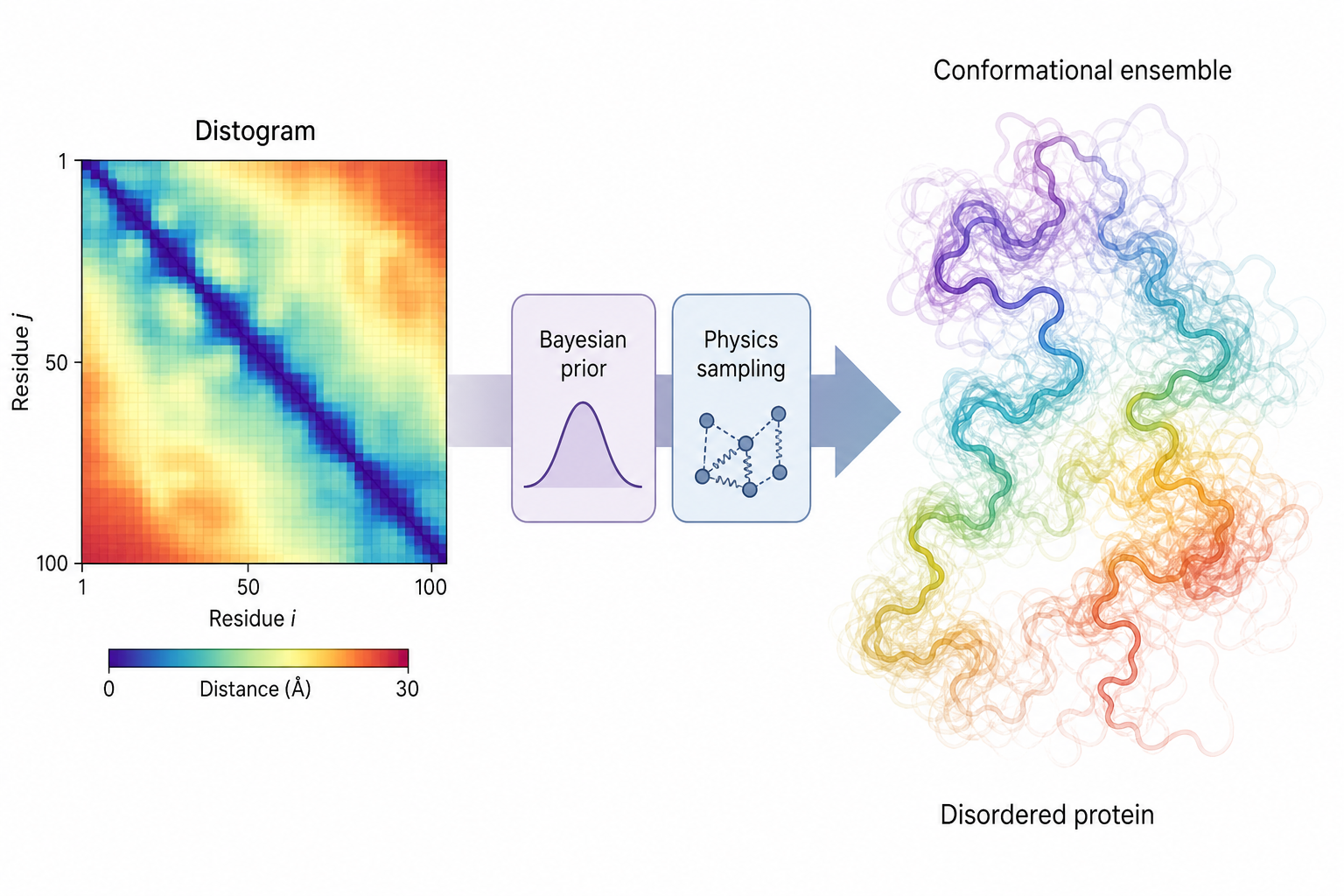
這張圖的生成提示詞(gpt-image-2, medium)
Left a colored residue-residue distance matrix "Distogram". Middle an arrow through "Bayesian prior + physics sampling". Right a flexible disordered protein chain as many transparent overlapping conformations "Conformational ensemble". English labels only: Distogram, Bayesian prior, Physics sampling, Conformational ensemble, Disordered protein. Elegant scientific infographic, soft gradients.
⑤ AlphaFold3 綜論 — 強大,但仍需 MD 補上動態
2025 年的綜論文章指出,AlphaFold3 等新版本在多鏈複合體、蛋白–RNA 和蛋白–配體(cofactor)方面有所突破,但仍難以描述活性位點的動態。
(此為綜述性結論,無對應實驗插圖;可參照本頁圖 1〜3 的互補概念。)
5突變如何透過結構與動態影響功能
把上述方法收斂到最常見的應用:單點突變
一個胺基酸的改變,可能改變側鏈方向、削弱與 DNA 的接觸、並提高局部柔性——結果就是結合變弱、穩定性改變。這正是 ESMDance、MELD-DNA 等方法想要量化預測的目標。
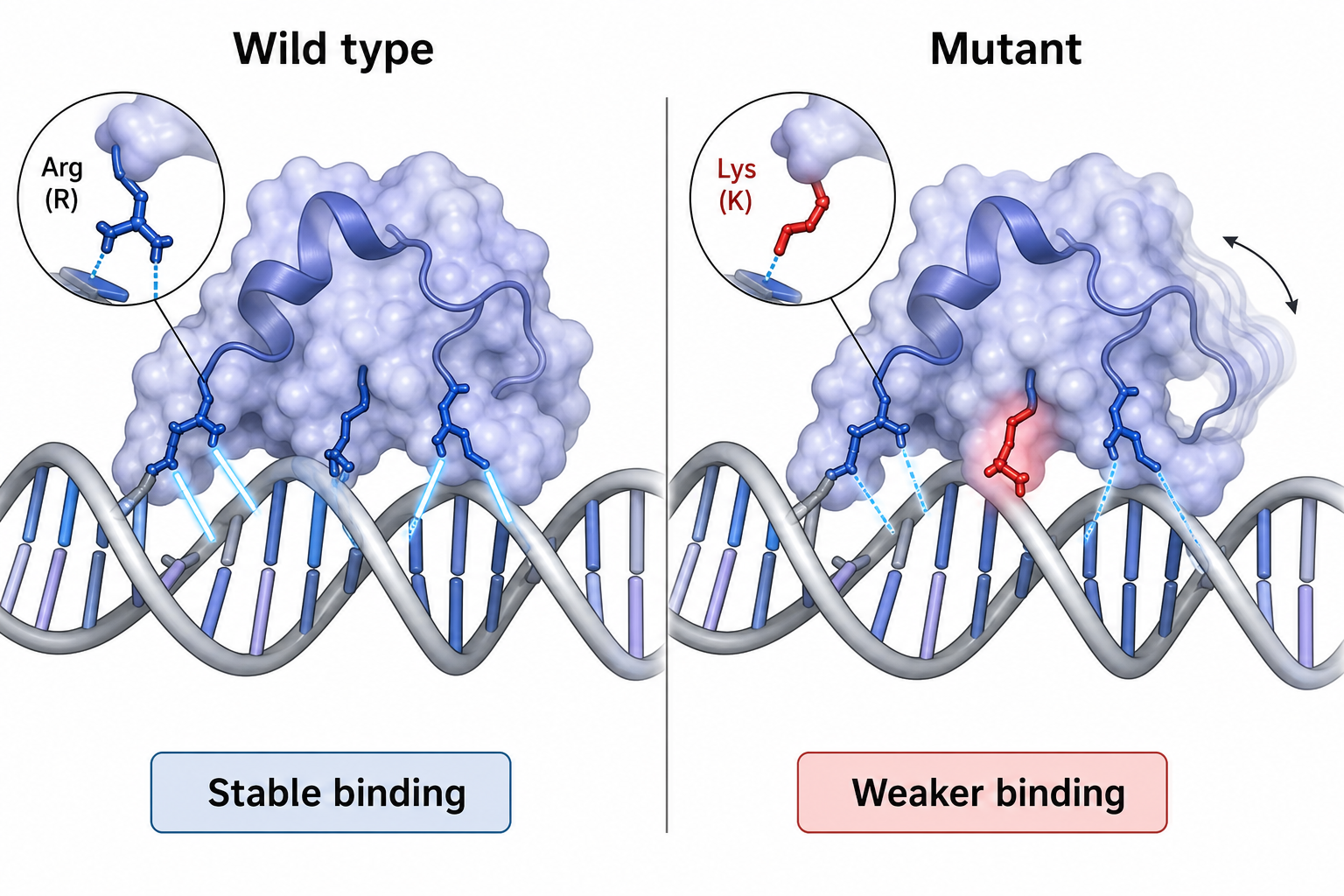
這張圖的生成提示詞(gpt-image-2, medium)
Compare wild-type and mutant protein binding to DNA. Left "Wild type" protein bound stably to a DNA helix with solid glowing contact lines. Right "Mutant" same protein with one residue changed (highlighted red), altered side chain, weakened dashed contacts, local flexibility as motion blur. Bottom labels "Stable binding" and "Weaker binding". Semi-realistic but simplified molecular biology style, English labels only.
6戰略意義與應用潛力
為什麼這套整合方法值得投入
- 彌補靜態模型的盲點:AlphaFold / ESM 主要捕捉序列與靜態結構,對無序區或缺乏同源序列的蛋白有限。把 MD 動態特徵納入訓練,可顯著提升突變效應預測準確度。
- 揭示新的功能位點:以 AF2 結構作為 MD 起點,能辨識新的結合斑塊與功能殘基——cohesin 研究即據此找到多個前未報導的 DNA 結合位點。
- 適用於資料稀缺或設計蛋白:SeqDance / ESMDance 顯示,依賴 MD 動態的模型在設計蛋白與病毒蛋白等缺乏同源信息時仍表現良好,對新蛋白設計特別有價值。
- 提升計算效率:bAIes 等方法利用 AF 距離分佈與簡化物理場,在不犧牲精度的前提下降低全原子 MD 的成本,對大量構象採樣的突變篩選與 IDP 研究尤為重要。
- 促進跨學科合作:對來自材料化學或其他領域的研究者,了解 MD 與深度學習模型的結合方法,可加速跨域的蛋白設計與功能預測,拓展研究方向。
7學生學習地圖
想自己動手,照這五階段往上走
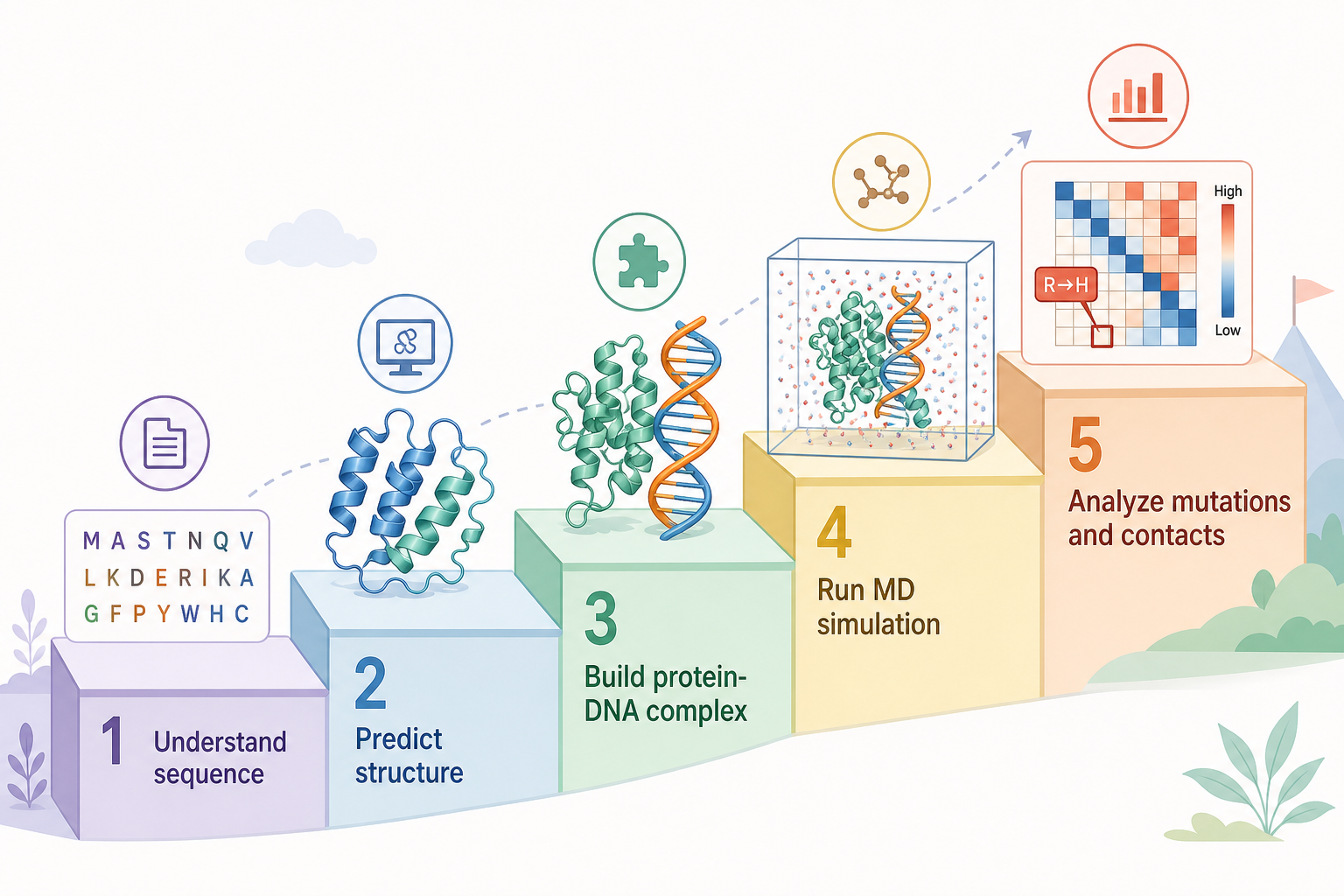
這張圖的生成提示詞(gpt-image-2, medium)
Roadmap as an ascending staircase with five steps for students. Step 1 "Understand sequence" amino acid letters; 2 "Predict structure" folded protein; 3 "Build protein-DNA complex" DNA helix; 4 "Run MD simulation" simulation box; 5 "Analyze mutations and contacts" heatmap + mutation marker. Small icon and short English label per step, approachable university teaching infographic, pastel colors.
8結語
From Sequence to Function
綜合上述研究可見,將 MD、AlphaFold、ESM 等工具結合,具有明確的科研與應用價值。這些方法既能補足靜態結構預測的不足,又能利用 MD 提供的動態資訊,預測突變效應與 DNA 結合能力;對於開發新型蛋白、解析機制、以及指導實驗設計,都具有戰略意義。
透過這些進展,研究者可以更全面地從序列出發,預測結構、動態與功能,促進蛋白工程與藥物研發。
§參考文獻
原綜述引用之 6 篇 PMC 文獻(可點擊)
- Protein Language Models Trained on Biophysical Dynamics Inform Mutation Effects(SeqDance / ESMDance)
PMC12846831 https://pmc.ncbi.nlm.nih.gov/articles/PMC12846831/ - Structural predictions of protein–DNA binding: MELD-DNA
PMC9976882 https://pmc.ncbi.nlm.nih.gov/articles/PMC9976882/ - Molecular dynamics simulations of human cohesin subunits identify DNA binding sites and their potential roles in DNA loop extrusion
PMC11970657 https://pmc.ncbi.nlm.nih.gov/articles/PMC11970657/ - Atomic resolution ensembles of intrinsically disordered proteins with AlphaFold(bAIes)
PMC12982490 https://pmc.ncbi.nlm.nih.gov/articles/PMC12982490/ - AlphaFold3: An Overview of Applications and Performance Insights
PMC12027460 https://pmc.ncbi.nlm.nih.gov/articles/PMC12027460/